Un ensayo clínico con medicamentos es una investigación que se realiza en personas, destinada a estudiar la eficacia y la seguridad de un fármaco. El objetivo de estos estudios es obtener datos que avalen que el balance beneficio/riesgo es positivo en una determinada terapia, y así poder solicitar posteriormente una autorización de comercialización para el medicamento.
Ya en los ensayos en Fase I. la población con la que se trabaja son pocos participantes (decenas), que pueden ser voluntarios sanos en función de la patología de estudio, y lo que realmente se estudia es confirmar que la administración del fármaco es segura.
En Fase II se amplía más la población a estudiar que puede llegar a las 200-300 personas, comienzan los análisis de eficacia siendo la seguridad con las dosis exploradas otro de los objetivos principales.
Continuando con los Fase III se busca la confirmación de la eficacia del fármaco en miles de participantes con el objetivo de obtener datos para una solicitud de autorización de comercialización y como objetivos secundarios se sigue ampliando la información de seguridad del fármaco a estudio. Finalmente, en los Fase IV son estudios a largo plazo con medicamentos ya autorizados para estudiar posibles reacciones adversas poco frecuentes o conocer posibles beneficios adicionales, u otras indicaciones.
¿Pero cómo se estudia realmente esa seguridad? Pues bien, en cualquier estudio en el que participes, estará el apartado en el cuaderno de recogida de datos de Eventos Adversos, esta información permitirá establecer una relación riesgo/beneficio, muy importante a la hora de someter un medicamento a registro sanitario.
Cuando comienzas a leer un protocolo de un ensayo clínico en sus primeras líneas, figura como propósito, objetivo primario o secundario explorar la seguridad del medicamento en la población a estudio. Uno de los objetivos fundamentales de un Estudio Clínico es conocer la seguridad del producto. Es por ello que los eventos adversos a medicamentos deben ser notificados.
Para ello y el propósito de estas líneas es definir y poner ejemplos de eventos adversos como documentarlos en la historia para que luego no nos falte información a la hora de registrarlo en el cuaderno de recogida de datos. ¿Comenzamos?
Acontecimiento Adverso (AA). Cualquier incidencia perjudicial para la salud que se presente en un paciente o sujeto de una investigación clínica al que se ha administrado un medicamento, aunque no tenga necesariamente una relación causal con dicho tratamiento. Un acontecimiento adverso (AA) puede ser, por tanto, cualquier signo desfavorable y no intencionado (incluyendo un hallazgo anormal de laboratorio), síntoma o enfermedad temporalmente asociada con el uso de un medicamento (en investigación), esté o no relacionado con el medicamento (en investigación).
En la mayoría de los protocolos los eventos adversos deben de notificarse desde la firma del ICF, aunque el paciente aun no esté recibiendo la medicación y hasta 30 días después de haberla detenido.
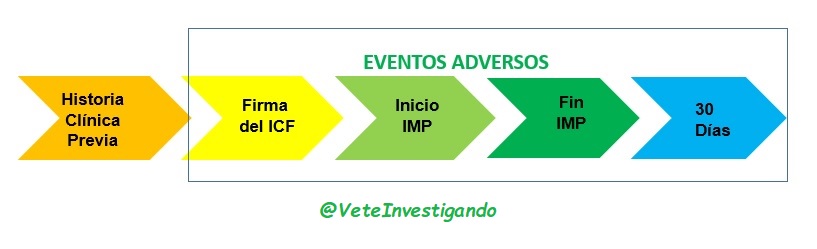
A la hora de documentarlo hay que registrar la siguiente información:
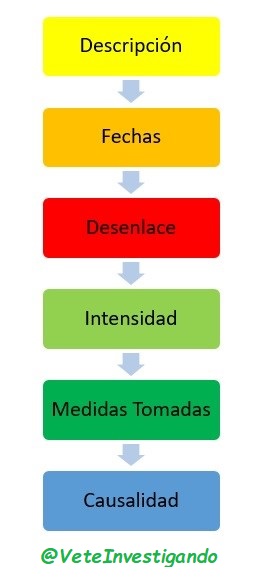
Ejemplo de Evento Adverso, puede ser que el paciente refiera malestar abdominal desde que está tomando el fármaco del estudio, o un simple resfriado, y habría que documentar cuando comenzó, si está tomando medicación para ello, que medicación, la dosis, la vía de administración y la frecuencia, y el desenlace de este acontecimiento adversos y si según el criterio del investigados este está o no relacionado con la medicación a estudio.

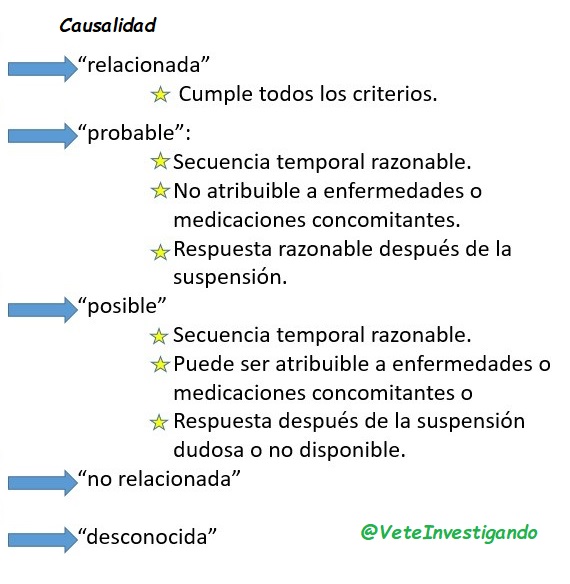
Reacción adversa: Es cualquier evento adverso en el que existe una relación causal claramente definida con un producto de investigación o existe al menos una posibilidad razonable de relación causal, que se presenta independientemente de la dosis administrada.
Es decir, si se considera que el evento adverso en cuestión está relacionado o existe esa posibilidad sería una reacción adversa, notificándose de igual manera en el cuaderno de recogida de datos.
Acontecimiento Adverso Grave (SAE) o Reacción Adversa Grave al Medicamento: Cualquier acontecimiento adverso o reacción adversa que a cualquier dosis:
• Produzca la muerte del paciente
• Amenace la vida del sujeto
• Haga necesaria la hospitalización del sujeto o la prolongación de ésta
• Produzca invalidez o incapacidad permanente o importante
• Dé lugar a una anomalía o malformación congénita.
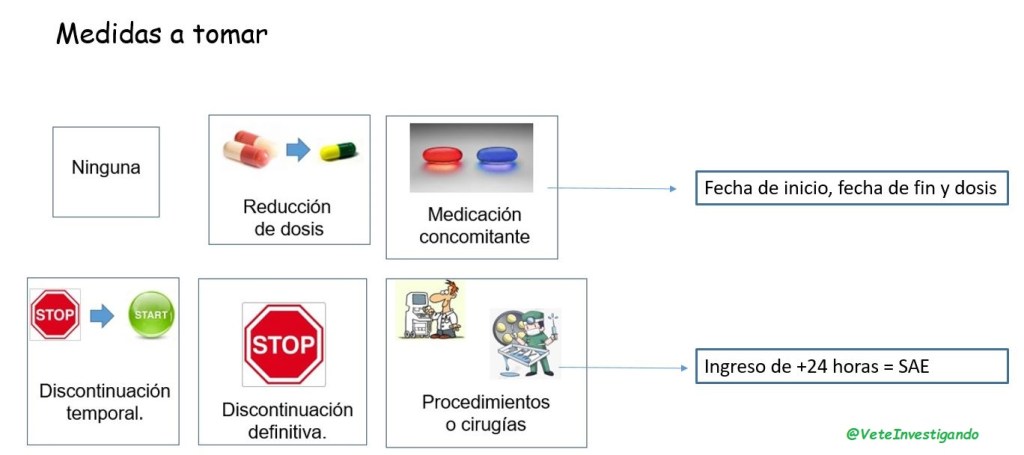
Se deberá notificar en las 24h posteriores a las que el investigador tenga conocimiento de ello y se hará igualmente en el cuaderno de recogida de datos, pero marcando en este caso en la intensidad GRAVE, y nos pedirá información adicional sobre este. También muchos de los ensayos aún siguen notificando los SAEs en papel y enviándolos por mail o fax.
Un ejemplo, sería una caída accidental con una fractura de cadera que requiera una cirugía con ingreso hospitalario de mas 24h. el cual no estaría relacionado con el producto en investigación, pero si cumpliría el criterio de gravedad por el ingreso.
Dentro de los SAEs también tenemos los embarazos, que deben de ser notificados y darles seguimiento.
Reacción adversa seria (RAS): Es cualquier reacción adversa que resulte en muerte, sea potencialmente mortal, requiera hospitalización o prolongación de la hospitalización, produzca discapacidad o incapacidad permanente o importante, provoque una anomalía o malformación congénita. A efectos de su notificación, se tratarán también como serios aquellos eventos que, desde el punto de vista médico pueden poner en peligro al sujeto de investigación o requerir una intervención para prevenir uno de los resultados señalados.
Aquí podríamos poner como ejemplo un trombo embolismo pulmonar, que requiera que el paciente acuda a urgencias y su ingreso hospitalario del paciente.
La diferencia clave entre evento y reacción es la relación de causalidad.
Evento adverso de interés especial (AESI): Un evento adverso de especial interés es un evento adverso que puede ser serio o no serio y podría incluir eventos que podrían ser precursores potenciales o pródromos de afecciones médicas más graves en personas susceptibles. Dichos eventos deben describirse en protocolos o enmiendas al protocolo, y deben proporcionarse instrucciones a los investigadores sobre cómo y cuándo deben informarse al patrocinador.
Dentro del propio protocolo vendrán marcados cuales se consideran AESI, un ejemplo que tuvimos fue en un ensayo que la diarrea pasaba a ser AESI, si las deposiciones diarias eran ≥5.
Sospecha de reacción adversa sería e Inesperada (RAGI) (SUSAR de sus siglas en Ingles): Es cualquier evento adverso serio en el que existe al menos una posibilidad razonable de relación causal con el producto en investigación y cuya naturaleza o gravedad no está descrita en el manual del investigador y/o ficha técnica.
Con respecto a los SUSAR cada ensayo los gestiona de diferente manera, les puede llegar una notificación al correo al IP (investigador principal) y él se encarga de distribuir la info a todo el equipo, hay que imprimirlo y firmarlo.
Otros ensayos tienen un portal donde te llega una notificación y debe el IP entrar y leer la info y darle como acknowledge. También hay veces que le llega un correo para descargarlo y puedes guardarlo electrónicamente esa info y distribuirla al equipo, y cada 6 meses también te llega un listado con todos los SUSAR que han surgido a modo de resumen.
Como podéis ver, es una información muy muy muy relevante, que debe documentar en todas y cada una de las visitas al centro de los pacientes, ya que dentro del “Scheduled assessment” siempre está la evaluación delos acontecimientos y adversos los cuales deben de preguntarse al paciente y esta informaci0n debe de ser monitorizada en todas las visitas de los monitores y revisar que toda la información este completa y correctamente documentada en el documento fuente, que es este caso siempre es la historia del paciente.