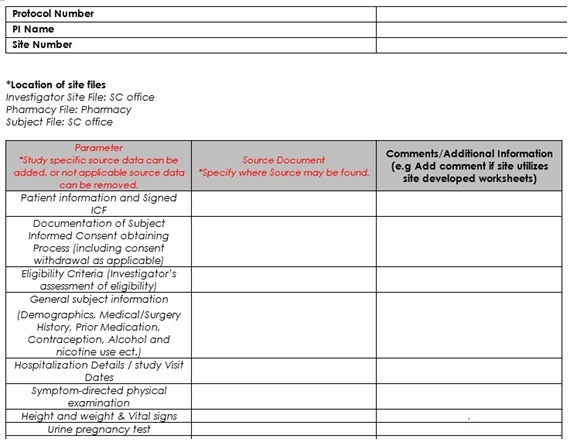
• Source data identification log. Todos los datos que se recogen en un ensayo clínico cuentan con un documento fuente donde se originan, con este log se intenta resumir cual es la localización original de cada dato o documento fuente. El termino de documento fuente se usa para describir la información original y copia certificadas de los datos, observaciones y otras actividades necesarias para la reconstrucción de la evaluación de una visita del ensayo. Estos documentos fuentes pueden ser: la historia clínica electrónica, las notas del laboratorio o los informes del laboratorio, los diarios de los pacientes, cuestionarios, los registros de dispensacióny administración de la medicación, radiografías, electros, tacs u otras pruebas de imagen, las notas de enfermería. Es importante registrarlo de manera clara para que poténciales auditores, monitores puedan localizar los datos fuente. Este log será completado el día de la visita de inicio previo al reclutamiento de los pacientes, y firmado por el investigador principal. Este también será guardado en el archivo del investigador.
• Site blinding plan: En los ensayos clínicos, el acto de cegar se refiere a mantener a una o más categorías de participantes sin conocimiento de la intervención asignada. Mantener «ciegos» a determinados participantes del estudio es importante para ayudar a reducir o eliminar los resultados sesgados. las normas internacionales definidas en la directriz de la ICH sobre Principios Estadísticos para Ensayos Clínicos (E9) afirman que «las técnicas más importantes para evitar el sesgo en los ensayos clínicos son la aleatorización y el cegamiento».
El propósito del blinding plan del centro es describir los métodos para mantener el ciego del estudio y evitar sesgos durante el ensayo, y describir las funciones de los miembros del equipo del estudio para los miembros del personal del centro ciegos y no ciegos.
Al principio del documento se hace una pequeña descripción del estudio y los grupos de tratamiento. Se especificara claramente la definición del personal ciego y no ciego, y a continuación se enumeran los miembros del equipo y la función que realizan dentro del estudio, detallando si son ciegos o no y los entrenamiento que ha recibido.
Debe de exponerse como se va a llevar a cabo el mantenimiento del ciego, justificando los puntos a tener en cuenta, por ejemplo:
La integridad del cegamiento del estudio se mantiene entre los miembros cegados y no cegados a nivel del centro, como se indica a continuación:
– Se mantiene entre los miembros cegados y los no cegados limitando el número de personal con acceso a la asignación del tratamiento.
– El investigador principal y el personal del estudio o sus delegados, así como los sujetos del estudio, estarán cegados a la asignación de los tratamientos farmacológicos del estudio.
– PNT del centro o plan de cegamiento que describa los procesos que garantizan el mantenimiento del cegamiento. El personal del centro debe confirmar la formación sobre los procedimientos del estudio ciego en el centro.
– Registro de delegación los farmacéuticos no ciegosy el personal delegado no ciego figuran en el registro de delegación del estudio como tales y son independientes de los demás miembros del equipo que realizan cualquier actividad relacionada con el estudio.
– IWRS y Gestión de IMP:
o Los sujetos serán aleatorizados en el IWRS y se proporcionará una notificación de confirmación ciega al personal ciego del estudio.
o El farmacéutico no ciego recibirá una notificación de aleatorización por correo electrónico con el número de kit de tratamiento que debe dispensar, sin información sobre la asignación al tratamiento DubiDubi frente al placebo.
o Todos los fármacos del estudio serán preparados y dispensados al equipo del centro por el farmacéutico no ciego designado o el personal del centro no ciego delegado.
Se describirá como se recibe, almacena, se devuelven, se cuenta la medicación, como viene empaquetada y en qué condiciones de, temperaturas y que hacer en caso de desviaciones de temperatura y quien tiene acceso a ella.
Habrá un apartado donde figurara como se restringen las comunicaciones entre el promotor y el centro para que vayan en la lista de personas no ciegos o ciegas según aplique y las visitas de los monitores.
Finalmente aparecerá un apartado donde se especifique como se puede hacer la rotura de ciego y que pasos hay que seguir.
Este documento debe de ser firmado al inicio del estudio por el Investigador principal.

Contabilidad de medicación.
Los investigadores por GCPs son responsables de mantener un control estricto sobre los fármacos en investigación para garantizar que el dispositivo se utiliza sólo para los sujetos inscritos en el estudio. El registro de dispensación/rendición de cuentas de fármacos o productos biológicos ayuda a mantener un inventario de los fármacos utilizados durante el estudio.
El ciclo de la medicación de los ensayos clínicos debe de seguir este esquema.
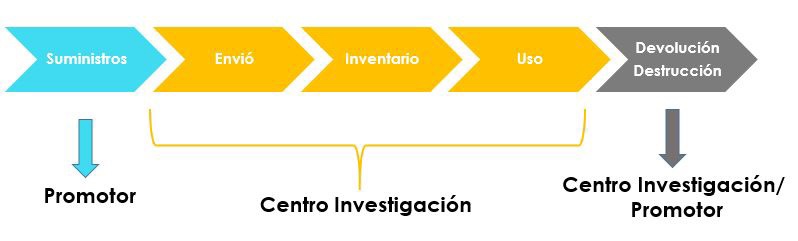
Cada ensayo clínico cuenta con sus propios log de contabilidad de medicación, y los centros pueden usarlos, o usan los suyos propios.
He aquí un ejemplo de un log de dispensación por parte de farmacia:
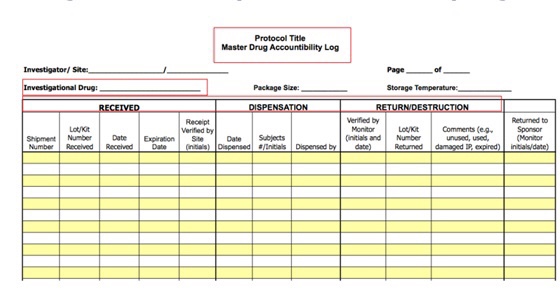
Donde se registra cuando se ha recibido, cuando se ha administrados y cuando ha sido devuelto y destruido (puede destruirla el centro después de la revisión del monitor o el propio promotor).
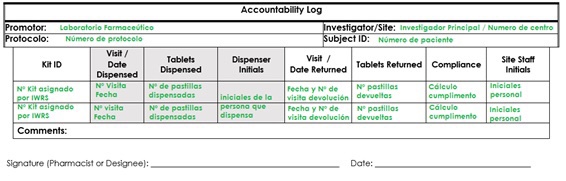
Desde el punto de vista de los coordinadores, tenemos otros logs que completar, puesto que como es mi caso, yo recojo la medicación de farmacia se la dispenso al paciente y este me la devuelve en visitas posteriores, para su contabilidad y registro de la adherencia del paciente al tratamiento del estudio.
Los registros de responsabilidad de medicamentos deben incluir:
Nombre del medicamento.
Número de kit dispensado.
La cantidad de fármaco recibida, utilizada, devuelta o desechada.
La cantidad de fármaco restante.
Los registros pueden requerir información adicional, según el tipo de ensayo y el tipo de fármaco. Por ejemplo, si el fármaco requiere preparación y se administra in situ, el registro debe incluir la fecha y la hora en que se administró. Si se prepara y el sujeto no se presenta, los centros deben registrar si se destruyó. Si el fármaco está refrigerado, debe registrarse la temperatura.
En muchos estudios, si el paciente no mantiene una adherencia al tratamiento superior a X % de la misma, se le discontinua del ensayo clínico.
Una vez que se sabe qué información debe incluirse, el siguiente paso es asegurarse de que se registra.
Si sus registros muestran que proporcionó 40 píldoras a un sujeto y él o ella de alguna manera tomó 42, eso es un problema. Puede que se trate de una simple cuestión matemática, pero garantizar una adecuada rendición de cuentas sobre los fármacos dista mucho de ser sencillo. Se requiere previsión y coherencia para hacerlo correctamente.
¿Qué os ha parecido la Versión 3.0?
¡Nos leemos en la siguiente!
